In 2002 we found that CEP-1347 is an inhibitor specific for Rac/CDC42-dependent kinases (PAKs/MLKs), and suppresses the growth of both RAS cancer and NF (neurofibromatosis) tumor cells where PAK1 is abnormally activated.
Interesting, like SAHA, CEP-1347 can pass through BBB (blood brain barrier) effectively so that it can reach brain tissues when this drug is administered systemically.
In 2003 Gillian Bates' group in London found that SAHA dramatically improves the motor impairment in HD mouse model (R6/2 mice). Like FK228, SAHA inhibits HDAC (histone deacetylase) directly, and eventually blocks the PAK1 signal pathways. However, neither FK228 nor SAHA was then available on the market. Besides, FK228 cannot cross the BBB.
In this fall Leslie Thomson’s group of UCI (University of California at Irvine) found that, like SAHA, CEP-1347 significantly reduces or delays the HD (Huntington’s Disease) symptom, both in vitro (cell culture) and in vivo (HD mouse model), suggesting that CEP-1347 and a few other anti-PAK1 drugs such as SAHA and Bio 30 could be potentially useful for the therapy of HD.
Since CEP-1347 is not available on the market as yet, it is likely that only two remedies so far available on the market, SAHA (expensive anti-CTCL drug) or Bio 30 (inexpensive healthcare product), would be used for the systemic (oral) treatment of HD patients at present. Like SAHA, Bio 30 can cross the BBB.
Mol Cell Neurosci. 2008 Sep;39: 8-20.
CEP-1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice.
Apostol BL, Simmons DA, Zuccato C, Illes K, Pallos J, Casale M, Conforti P, Ramos C, Roarke M, Kathuria S, Cattaneo E, Marsh JL, Thompson LM.
Department of Psychiatry and Human Behavior, University of California, Irvine, CA 92697, USA.
Huntington's disease (HD) is a devastating neurodegenerative disorder caused by an expanded polyglutamine repeat within the protein Huntingtin (Htt). We previously reported that mutant Htt expression activates the ERK1/2 and JNK pathways [Apostol, B.L.et al, 2006]. Chemical and genetic modulation of these pathways promotes cell survival and death, respectively.
Here we test the ability of two closely related compounds, CEP-11004 and CEP-1347, which inhibit Mixed Lineage Kinases (MLKs) and are neuroprotective, to suppress mutant Htt-mediated pathogenesis in multiple model systems. CEP-11004/CEP-1347 treatment significantly decreased toxicity in mutant Htt-expressing cells that evoke a strong JNK response. However, suppression of cellular dysfunction in cell lines that exhibit only mild Htt-associated toxicity and little JNK activation was associated with activation of ERK1/2. These compounds also reduced neurotoxicity in immortalized striatal neurons from mutant knock-in mice and Drosophila expressing a mutant Htt fragment.
Finally, CEP-1347 improved motor performance in R6/2 mice and restored expression of BDNF, a critical neurotrophic factor that is reduced in HD. These studies suggest a novel therapeutic approach for a currently untreatable neurodegenerative disease, HD, via CEP-1347 up-regulation of BDNF.
2008年9月23日火曜日
2008年9月22日月曜日
Mesothelioma: A new oncogenic target (YAP1)
of the tumor suppressor “Merlin”
In 2004 we found that Merlin, an NF2 gene product, directly inhibits the oncogenic kinase PAK1, and anti-PAK1 drugs such as CEP-1347 and FK228 suppress the growth of NF2-deficient mesothelioma and Schwannoma cell lines.
In summer of 2008, Yoshitaka Sekido’s group at Aichi Cancer Center, Japan, found another target of tumor suppressor “Merlin”, which is also an oncogene product of 65 kDa called YAP1 (YES-associated protein 1). YAP1 is active as a transcription-coactivator (activating a p53-related transcription factor called p73) only when it is localized in nucleus. However, “Merlin” causes the phosphorylation of YAP1 at Ser 127 by the kinase LATS1, and blocks the nuclear localization of YAP1. Thus, “Merlin” is the negative regulator of two oncoproteins, YAP1 and PAK1.
It should be worth noting that unlike PAK1, YAP1 does not bind "Merlin" directly, and nobody knows how "Merlin" activates LATS1. It is possible that PAK1 normally inactivate LATS1, and "Merlin" simply reverses the action of PAK1...
Interestingly, in malignant pleural mesotheliomas (MPMs), YAP1 gene is often over-expressed, and NF2 gene is often deleted (or dysfunctions). This could be explained by the fact that "Merlin" inactivates YAP1 gene somehow.
Since we discovered that “Bio 30”, a CAPE-rich water-miscible extract of propolis from NZ (New Zealand) , suppresses the growth of NF2-deficient Schwannoma cells both in vitro (cell culture) and in vivo (xenograft in mice) by blocking the oncogenic PAK1 pathways, it would be of great interest to examine whether like “Merlin”, Bio 30 causes the phosphorylation of YAP1 at Ser 127, inactivating YAP1. If so, since YAP1 is essential for the growth of these MPMs, Bio 30 could be useful for the treatment of the formidable MPMs, in addition to brain tumors such as NF (neurofibromatosis) and gliomas as well as pancreatic and breast cancers.
Carcinogenesis (PubMed, 2008 Aug 25):
YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation.
Yokoyama T, Osada H, Murakami H, Tatematsu Y, Taniguchi T, Kondo Y, Yatabe Y, Hasegawa Y, Shimokata K, Horio Y, Hida T, Sekido Y.
Division of Molecular Oncology, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan.
We previously reported the results of BAC-array-CGH of malignant pleural mesotheliomas (MPMs), including two cases with high-level amplification in the 11q22 locus. In this study, we found that the YAP1 gene encoding a transcriptional co-activator, was localized in this amplified region and overexpressed in both cases, suggesting it as a candidate oncogene in this region. We analyzed the involvement of YAP1 in MPM proliferation, as well as its functional and physical interaction with Merlin encoded by the neurofibromatosis type 2 (NF2) tumor-suppressor gene, which is frequently mutated in MPMs. YAP1-RNAi suppressed growth of a mesothelioma cell line NCI-H290 with NF2 homozygous deletion, probably through cell-cycle arrest and apoptosis induction, while YAP1 transfection promoted the growth of MeT-5A, an immortalized mesothelial cell line. We also found that the introduction of NF2 into NCI-H290 induced phosphorylation at Ser127 of YAP1, which was accompanied by reduction of nuclear localization of YAP1, whereas nuclear localization of a YAP1 S127A-mutant was not affected. Furthermore, results of immunoprecipitation and in vitro pull-down assays indicated a physical interaction between Merlin and YAP1. These results suggest that YAP1 is involved in mesothelial cell growth, and that the transcriptional co-activator activity of YAP1 is functionally inhibited by Merlin through the induction of phosphorylation and cytoplasmic retention of YAP1. This is the first report of negative-regulatory signaling from Merlin to YAP1 in mammalian cells. Future studies of transcriptional targets of YAP1 in MPMs may shed light on the molecular mechanisms of MPM development and lead to new therapeutic strategies.
In summer of 2008, Yoshitaka Sekido’s group at Aichi Cancer Center, Japan, found another target of tumor suppressor “Merlin”, which is also an oncogene product of 65 kDa called YAP1 (YES-associated protein 1). YAP1 is active as a transcription-coactivator (activating a p53-related transcription factor called p73) only when it is localized in nucleus. However, “Merlin” causes the phosphorylation of YAP1 at Ser 127 by the kinase LATS1, and blocks the nuclear localization of YAP1. Thus, “Merlin” is the negative regulator of two oncoproteins, YAP1 and PAK1.
It should be worth noting that unlike PAK1, YAP1 does not bind "Merlin" directly, and nobody knows how "Merlin" activates LATS1. It is possible that PAK1 normally inactivate LATS1, and "Merlin" simply reverses the action of PAK1...
Interestingly, in malignant pleural mesotheliomas (MPMs), YAP1 gene is often over-expressed, and NF2 gene is often deleted (or dysfunctions). This could be explained by the fact that "Merlin" inactivates YAP1 gene somehow.
Since we discovered that “Bio 30”, a CAPE-rich water-miscible extract of propolis from NZ (New Zealand) , suppresses the growth of NF2-deficient Schwannoma cells both in vitro (cell culture) and in vivo (xenograft in mice) by blocking the oncogenic PAK1 pathways, it would be of great interest to examine whether like “Merlin”, Bio 30 causes the phosphorylation of YAP1 at Ser 127, inactivating YAP1. If so, since YAP1 is essential for the growth of these MPMs, Bio 30 could be useful for the treatment of the formidable MPMs, in addition to brain tumors such as NF (neurofibromatosis) and gliomas as well as pancreatic and breast cancers.
Carcinogenesis (PubMed, 2008 Aug 25):
YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation.
Yokoyama T, Osada H, Murakami H, Tatematsu Y, Taniguchi T, Kondo Y, Yatabe Y, Hasegawa Y, Shimokata K, Horio Y, Hida T, Sekido Y.
Division of Molecular Oncology, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan.
We previously reported the results of BAC-array-CGH of malignant pleural mesotheliomas (MPMs), including two cases with high-level amplification in the 11q22 locus. In this study, we found that the YAP1 gene encoding a transcriptional co-activator, was localized in this amplified region and overexpressed in both cases, suggesting it as a candidate oncogene in this region. We analyzed the involvement of YAP1 in MPM proliferation, as well as its functional and physical interaction with Merlin encoded by the neurofibromatosis type 2 (NF2) tumor-suppressor gene, which is frequently mutated in MPMs. YAP1-RNAi suppressed growth of a mesothelioma cell line NCI-H290 with NF2 homozygous deletion, probably through cell-cycle arrest and apoptosis induction, while YAP1 transfection promoted the growth of MeT-5A, an immortalized mesothelial cell line. We also found that the introduction of NF2 into NCI-H290 induced phosphorylation at Ser127 of YAP1, which was accompanied by reduction of nuclear localization of YAP1, whereas nuclear localization of a YAP1 S127A-mutant was not affected. Furthermore, results of immunoprecipitation and in vitro pull-down assays indicated a physical interaction between Merlin and YAP1. These results suggest that YAP1 is involved in mesothelial cell growth, and that the transcriptional co-activator activity of YAP1 is functionally inhibited by Merlin through the induction of phosphorylation and cytoplasmic retention of YAP1. This is the first report of negative-regulatory signaling from Merlin to YAP1 in mammalian cells. Future studies of transcriptional targets of YAP1 in MPMs may shed light on the molecular mechanisms of MPM development and lead to new therapeutic strategies.
2008年9月18日木曜日
Bio 30 Blocks the Growth of Deadly Gliomas
Bio 30, a CAPE-rich water-miscible extract of propolis from NZ (New Zealand),
has been shown by us (Demestre, M. et al, 2008) to suppress the growth of several tumors in vivo (xenografts in mice): deadly pancreatic cancer, breast cancer, and NF (neurofibromatosis) tumors (both deadly NF1 MPNST and NF2 Schwannoma).
Several years ago David Kaplan's group in Canada found that the GTPase Rac is essential for the growth of glioma cells (Senger, D. et al, 2002). Interestingly, CAPE down-regulates Rac, eventually inactivating the oncogenic kinase PAK1.
Thus, we recently tested the therapeutic effect of Bio 30 (100-300 mg/kg,
i.p., twice a week) on human grade 4 glioma (U87MG) xenograft in mice over
a few weeks. Both doses of Bio 30 turned out to be sufficient for the
complete suppression of glioma growth (Messerli, S. et al, 2008). We continue testing if Bio 30 also causes any regression of this deadly tumor.
Anti-cancer ingredients such as CAPE (caffeic acid phenethyl ester) in Bio
30 can easily pass the BBB (blood brain barrier), and CAPE alone has been shown by a Taiwanese group to suppress the growth of glioma (C6) xenograft in mice a few years ago (Kuo, HG et al, 2006).
However, CAPE has never been used clinically (and is not available on the market), because of its poor bioavailability. Unlike CAPE alone, Bio 30 has a very good bioavailabity, because it contains a plenty of lipids which solubilize CAPE and several other water-insoluble anti-cancer ingredients. Besides, like many other propolis, Bio 30 can boost the immune system to suppress the growth of cancers in general. Thus, Bio 30 would be potentially good for the systemic treatment of brain tumors such as NF and gliomas.
So far no effective therapeutic for glioma is available on the market. Thus,
it is likely that Bio 30 (alcohol-free liquid) from Manuka Health, NZ would serve as the first effective therapeutic for gliomas. It is both very safe and inexpensive
(costing only a dollar for the daily treatment). The recommended daily dose
would be 1 ml per 10 kg (bodyweight), meaning 6 ml per day for patients weighing
60 kg (130-135 pounds) who suffer from glioma or other PAK1-dependent tumors
such as NF and pancreatic cancers.
Continued
has been shown by us (Demestre, M. et al, 2008) to suppress the growth of several tumors in vivo (xenografts in mice): deadly pancreatic cancer, breast cancer, and NF (neurofibromatosis) tumors (both deadly NF1 MPNST and NF2 Schwannoma).
Several years ago David Kaplan's group in Canada found that the GTPase Rac is essential for the growth of glioma cells (Senger, D. et al, 2002). Interestingly, CAPE down-regulates Rac, eventually inactivating the oncogenic kinase PAK1.
Thus, we recently tested the therapeutic effect of Bio 30 (100-300 mg/kg,
i.p., twice a week) on human grade 4 glioma (U87MG) xenograft in mice over
a few weeks. Both doses of Bio 30 turned out to be sufficient for the
complete suppression of glioma growth (Messerli, S. et al, 2008). We continue testing if Bio 30 also causes any regression of this deadly tumor.
Anti-cancer ingredients such as CAPE (caffeic acid phenethyl ester) in Bio
30 can easily pass the BBB (blood brain barrier), and CAPE alone has been shown by a Taiwanese group to suppress the growth of glioma (C6) xenograft in mice a few years ago (Kuo, HG et al, 2006).
However, CAPE has never been used clinically (and is not available on the market), because of its poor bioavailability. Unlike CAPE alone, Bio 30 has a very good bioavailabity, because it contains a plenty of lipids which solubilize CAPE and several other water-insoluble anti-cancer ingredients. Besides, like many other propolis, Bio 30 can boost the immune system to suppress the growth of cancers in general. Thus, Bio 30 would be potentially good for the systemic treatment of brain tumors such as NF and gliomas.
So far no effective therapeutic for glioma is available on the market. Thus,
it is likely that Bio 30 (alcohol-free liquid) from Manuka Health, NZ would serve as the first effective therapeutic for gliomas. It is both very safe and inexpensive
(costing only a dollar for the daily treatment). The recommended daily dose
would be 1 ml per 10 kg (bodyweight), meaning 6 ml per day for patients weighing
60 kg (130-135 pounds) who suffer from glioma or other PAK1-dependent tumors
such as NF and pancreatic cancers.
Continued
2008年9月15日月曜日
2008 "Lasker" Award for Clinical Research
http://www.laskerfoundation.org/awards/2008_c_description.htm
Winner: Akira Endo (74)
For the discovery of the statins — drugs with remarkable LDL-cholesterol-lowering properties that have revolutionized the prevention and treatment of coronary heart disease.
The 2008 Lasker~DeBakey Award for Clinical Medical Research honors a scientist who discovered statins—drugs with remarkable LDL-cholesterol-lowering properties that have revolutionized the prevention and treatment of coronary heart disease (CHD). Akira Endo (Biopharm Research Laboratories, Inc., Tokyo) sifted through thousands of organisms, hunting for natural substances that block a key enzyme in the biochemical pathway that produces cholesterol, a major contributor to CHD. Remarkably, the compound that Endo found lowers concentrations of LDL (the bad cholesterol) but not HDL (the good cholesterol) in the bloodstream of animals and humans. His work stimulated Merck, Sharp & Dohme Research Laboratories (Merck) to launch a drug-development program that led, 20 years ago, to the first statin approved for medical use. This advance paved a path for other pharmaceutical companies to follow.
LDL in the bloodstream can form fatty deposits that narrow blood vessels. When this process occurs in arteries that deliver blood to the heart, it can lead to CHD, the major cause of chest pain and heart attack and the top killer in the industrialized world. In the US, more than 450,000 people died from CHD in 2004. In 2005, the American Heart Association estimated that 16 million Americans had CHD and 1.2 million would have a new or recurrent heart attack that year. Statin use is increasing—30 million people worldwide are taking them—and has begun to make a dent in those numbers. The drugs dramatically reduce the risk of CHD and its associated life-threatening events. Furthermore, studies with statins have erased long-standing doubt about the possibility of safely reducing cholesterol quantities with pharmaceutical agents.
Promising Early Results
In the 1950s and 1960s, epidemiological studies suggested a link between LDL cholesterol in the blood and CHD. Reducing LDL would shrink the incidence of this illness, some experts reasoned. However, many scientists and clinicians questioned whether doing so would stir trouble. Cholesterol is a crucial component of the membranes that encase our cells, and it serves as a raw material for other essential molecules, including some hormones and the sheath that insulates nerves. Further exacerbating uncertainty, most early agents that reduced cholesterol quantities in the blood cut concentrations only modestly and triggered unwelcome side effects. Therefore, skepticism simmered about the wisdom of lowering cholesterol concentrations.
Despite the concerns, efforts pushed forward. Humans acquire cholesterol from food and we also make it in our bodies, mostly in the liver. Dietary interventions to limit cholesterol intake had met with poor success and the idea of short-circuiting our ability to produce the compound seemed attractive. An enzyme called HMG-CoA reductase plays a central role in the manufacture of cholesterol. In the biochemical pathway that generates cholesterol, it converts a precursor molecule, HMG-CoA, into the next compound, mevalonate.
By 1971, when Endo began his work (at Sankyo Company in Tokyo), thwarting the reductase seemed like a possible way to keep the body's cholesterol production in check. Endo had a novel idea about how to find substances that block the reductase. Aware that organisms can secrete compounds with powerful biological activities—presumably to kill their competitors—Endo proposed that some creatures might spit out chemicals that foil the reductase and thus impede a vital activity of their neighbors.
Over the next two years, Endo and colleagues grew more than 6000 fungi, harvested the broth in which each had grown, and tested whether the material could interfere with an early step of cholesterol synthesis in a test tube. They then separated its components from one another, keeping track of the active material. By this and additional methods, he purified a substance from the fungus Penicillium citrinum, called mevastatin or "compactin", that blocks the reductase.
Additional analysis revealed that compactin resembles HMG-CoA and competes strongly with it for the site on the enzyme that catalyzes HMG-CoA's conversion to mevalonate. This capacity to bind specifically to the spot normally reserved for the enzyme's substrate suggested that the potential drug would not randomly clasp molecules in cells and disrupt other important activities.
Endo wondered whether compactin would reduce enzyme activity in animals, as it does in test tubes. In 1979, he showed that the compound dramatically lowers blood cholesterol in dogs and monkeys with no obvious toxic effects.
While these results were coming in, Endo and physician Akira Yamamoto (National Cardiovascular Center in Osaka) gave compactin to patients with extremely high LDL-cholesterol levels. In 1980, they reported that it cut LDL in the blood by an average of 27%. The next year, Hiroshi Mabuchi of Kanazawa School of Medicine published similarly promising findings. Furthermore, he established that the compound did not perturb quantities of a molecule that is involved in the cell's energy-production system whose concentrations experts thought might drop when HMG-CoA activity was stymied. These observations fed hope that compactin could reduce cholesterol without causing harm.
At the same time, different researchers were demonstrating the mechanism by which the drug acted, which provided a rationale for how it could operate safely. In 1974, Michael Brown and Joseph Goldstein (Lasker Basic Medical Research Award and Nobel Prize, 1985) at the University of Texas Southwestern Medical School discovered that cell-surface molecules known as LDL receptors on liver cells grab LDL in the blood. In 1981, they showed that, when statins lower reductase activity and cholesterol production wanes, cells place additional LDL receptors on their surfaces. Statins thus harness a normal mechanism by which cells drain cholesterol from the blood, yet ensure a steady supply of the molecule internally for vital activities.
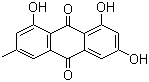
Statins reducing cholesterol:
Compactin (C), the first statin, discovered by Akira Endo;
Lovastatin (L), the first statin approved for clinical use, developed by Merck.
the compound "L" is a methylated form of the compound "C".
Statins in the Clinic
In 1978, Endo left Sankyo and joined the faculty of Tokyo Noko University. There, he isolated additional HMG-CoA reductase inhibitors, including one called monacolin K, from another fungus. Merck scientists, led by Alfred W. Alberts, independently identified—from a different organism—the same compound, which they called mevinolin (and subsequently renamed "lovastatin"). The company had been pursuing statin-related research since soon after Endo published his first papers in 1976. At that time, P. Roy Vagelos, then President of Merck Research Laboratories, had signed an agreement with Sankyo and obtained samples of compactin from Endo, with which Merck confirmed Endo's results and launched its own program.
In April 1980, Merck began clinical studies of lovastatin. Several months later, rumors emerged from Sankyo that the drug caused tumors in dogs. Merck halted its studies. However, experts in the field urged the company forward. The dogs were receiving exceptionally high doses and Sankyo provided no data for others to assess. Investigators were witnessing dramatic benefits with compactin, particularly to individuals who have a genetic alteration in one of their two copies of the gene for the LDL receptor and thus carry colossal quantities of circulating cholesterol; these patients are extremely difficult to treat.
Several years later, Merck picked up its clinical trials on lovastatin. In 1987, the FDA approved the drug (Mevacor®).
The success of compactin and lovastatin inspired efforts to improve the statins by chemically modifying the natural compounds or crafting synthetic ones. Because the statins curb LDL so much more than any of the existing treatments, it became possible to demonstrate unequivocally that the drugs decrease CHD events without serious side effects. For example, in 1994, the Scandinavian Simvastatin Survival Study (4S study) showed that treatment with simvastatin (the second Merck statin) —for as short a period as five years—lowered LDL levels by 35%; this reduction was associated with a drop of 40% in CHD deaths. A 2005 analysis of more than 90,000 people in multiple 5-year randomized studies obtained similar results. Furthermore, this work established that people reap these benefits without an increase in cancer, other diseases, or deaths from any cause.
Statins are now routinely used to prevent and treat CHD throughout the world. Although CHD is aggravated by multiple risk factors in addition to high LDL—cigarette smoking, high blood pressure, obesity, and diabetes —reducing LDL levels alone makes a significant impact.
By discovering statins, Endo ushered in a new era in preventing and treating CHD, the leading cause of death in the US and a major source of human suffering. His work has touched millions of people and promises to prolong and improve the lives of many millions more.
by Evelyn Strauss, Ph.D.
Winner: Akira Endo (74)
For the discovery of the statins — drugs with remarkable LDL-cholesterol-lowering properties that have revolutionized the prevention and treatment of coronary heart disease.
The 2008 Lasker~DeBakey Award for Clinical Medical Research honors a scientist who discovered statins—drugs with remarkable LDL-cholesterol-lowering properties that have revolutionized the prevention and treatment of coronary heart disease (CHD). Akira Endo (Biopharm Research Laboratories, Inc., Tokyo) sifted through thousands of organisms, hunting for natural substances that block a key enzyme in the biochemical pathway that produces cholesterol, a major contributor to CHD. Remarkably, the compound that Endo found lowers concentrations of LDL (the bad cholesterol) but not HDL (the good cholesterol) in the bloodstream of animals and humans. His work stimulated Merck, Sharp & Dohme Research Laboratories (Merck) to launch a drug-development program that led, 20 years ago, to the first statin approved for medical use. This advance paved a path for other pharmaceutical companies to follow.
LDL in the bloodstream can form fatty deposits that narrow blood vessels. When this process occurs in arteries that deliver blood to the heart, it can lead to CHD, the major cause of chest pain and heart attack and the top killer in the industrialized world. In the US, more than 450,000 people died from CHD in 2004. In 2005, the American Heart Association estimated that 16 million Americans had CHD and 1.2 million would have a new or recurrent heart attack that year. Statin use is increasing—30 million people worldwide are taking them—and has begun to make a dent in those numbers. The drugs dramatically reduce the risk of CHD and its associated life-threatening events. Furthermore, studies with statins have erased long-standing doubt about the possibility of safely reducing cholesterol quantities with pharmaceutical agents.
Promising Early Results
In the 1950s and 1960s, epidemiological studies suggested a link between LDL cholesterol in the blood and CHD. Reducing LDL would shrink the incidence of this illness, some experts reasoned. However, many scientists and clinicians questioned whether doing so would stir trouble. Cholesterol is a crucial component of the membranes that encase our cells, and it serves as a raw material for other essential molecules, including some hormones and the sheath that insulates nerves. Further exacerbating uncertainty, most early agents that reduced cholesterol quantities in the blood cut concentrations only modestly and triggered unwelcome side effects. Therefore, skepticism simmered about the wisdom of lowering cholesterol concentrations.
Despite the concerns, efforts pushed forward. Humans acquire cholesterol from food and we also make it in our bodies, mostly in the liver. Dietary interventions to limit cholesterol intake had met with poor success and the idea of short-circuiting our ability to produce the compound seemed attractive. An enzyme called HMG-CoA reductase plays a central role in the manufacture of cholesterol. In the biochemical pathway that generates cholesterol, it converts a precursor molecule, HMG-CoA, into the next compound, mevalonate.
By 1971, when Endo began his work (at Sankyo Company in Tokyo), thwarting the reductase seemed like a possible way to keep the body's cholesterol production in check. Endo had a novel idea about how to find substances that block the reductase. Aware that organisms can secrete compounds with powerful biological activities—presumably to kill their competitors—Endo proposed that some creatures might spit out chemicals that foil the reductase and thus impede a vital activity of their neighbors.
Over the next two years, Endo and colleagues grew more than 6000 fungi, harvested the broth in which each had grown, and tested whether the material could interfere with an early step of cholesterol synthesis in a test tube. They then separated its components from one another, keeping track of the active material. By this and additional methods, he purified a substance from the fungus Penicillium citrinum, called mevastatin or "compactin", that blocks the reductase.
Additional analysis revealed that compactin resembles HMG-CoA and competes strongly with it for the site on the enzyme that catalyzes HMG-CoA's conversion to mevalonate. This capacity to bind specifically to the spot normally reserved for the enzyme's substrate suggested that the potential drug would not randomly clasp molecules in cells and disrupt other important activities.
Endo wondered whether compactin would reduce enzyme activity in animals, as it does in test tubes. In 1979, he showed that the compound dramatically lowers blood cholesterol in dogs and monkeys with no obvious toxic effects.
While these results were coming in, Endo and physician Akira Yamamoto (National Cardiovascular Center in Osaka) gave compactin to patients with extremely high LDL-cholesterol levels. In 1980, they reported that it cut LDL in the blood by an average of 27%. The next year, Hiroshi Mabuchi of Kanazawa School of Medicine published similarly promising findings. Furthermore, he established that the compound did not perturb quantities of a molecule that is involved in the cell's energy-production system whose concentrations experts thought might drop when HMG-CoA activity was stymied. These observations fed hope that compactin could reduce cholesterol without causing harm.
At the same time, different researchers were demonstrating the mechanism by which the drug acted, which provided a rationale for how it could operate safely. In 1974, Michael Brown and Joseph Goldstein (Lasker Basic Medical Research Award and Nobel Prize, 1985) at the University of Texas Southwestern Medical School discovered that cell-surface molecules known as LDL receptors on liver cells grab LDL in the blood. In 1981, they showed that, when statins lower reductase activity and cholesterol production wanes, cells place additional LDL receptors on their surfaces. Statins thus harness a normal mechanism by which cells drain cholesterol from the blood, yet ensure a steady supply of the molecule internally for vital activities.
Statins reducing cholesterol:
Compactin (C), the first statin, discovered by Akira Endo;
Lovastatin (L), the first statin approved for clinical use, developed by Merck.
the compound "L" is a methylated form of the compound "C".
Statins in the Clinic
In 1978, Endo left Sankyo and joined the faculty of Tokyo Noko University. There, he isolated additional HMG-CoA reductase inhibitors, including one called monacolin K, from another fungus. Merck scientists, led by Alfred W. Alberts, independently identified—from a different organism—the same compound, which they called mevinolin (and subsequently renamed "lovastatin"). The company had been pursuing statin-related research since soon after Endo published his first papers in 1976. At that time, P. Roy Vagelos, then President of Merck Research Laboratories, had signed an agreement with Sankyo and obtained samples of compactin from Endo, with which Merck confirmed Endo's results and launched its own program.
In April 1980, Merck began clinical studies of lovastatin. Several months later, rumors emerged from Sankyo that the drug caused tumors in dogs. Merck halted its studies. However, experts in the field urged the company forward. The dogs were receiving exceptionally high doses and Sankyo provided no data for others to assess. Investigators were witnessing dramatic benefits with compactin, particularly to individuals who have a genetic alteration in one of their two copies of the gene for the LDL receptor and thus carry colossal quantities of circulating cholesterol; these patients are extremely difficult to treat.
Several years later, Merck picked up its clinical trials on lovastatin. In 1987, the FDA approved the drug (Mevacor®).
The success of compactin and lovastatin inspired efforts to improve the statins by chemically modifying the natural compounds or crafting synthetic ones. Because the statins curb LDL so much more than any of the existing treatments, it became possible to demonstrate unequivocally that the drugs decrease CHD events without serious side effects. For example, in 1994, the Scandinavian Simvastatin Survival Study (4S study) showed that treatment with simvastatin (the second Merck statin) —for as short a period as five years—lowered LDL levels by 35%; this reduction was associated with a drop of 40% in CHD deaths. A 2005 analysis of more than 90,000 people in multiple 5-year randomized studies obtained similar results. Furthermore, this work established that people reap these benefits without an increase in cancer, other diseases, or deaths from any cause.
Statins are now routinely used to prevent and treat CHD throughout the world. Although CHD is aggravated by multiple risk factors in addition to high LDL—cigarette smoking, high blood pressure, obesity, and diabetes —reducing LDL levels alone makes a significant impact.
By discovering statins, Endo ushered in a new era in preventing and treating CHD, the leading cause of death in the US and a major source of human suffering. His work has touched millions of people and promises to prolong and improve the lives of many millions more.
by Evelyn Strauss, Ph.D.
登録:
投稿 (Atom)









































































![リオ五輪男子体操団体:日本(金)、ロシア[銀]、中国[銅]。](https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEjHS61FORcH43CteZVfJzLmbqvNwOIliOSMpTpRtEi7x8j1ZwPk5rDaZovTrwuZxfDDtdEDSj673it735LF0mweIunaj7ja07lURBDYTV6wPMaAlumFt3aWWzYbHZgIaxcOLk_OKEMyQ3lX/s1600/2016+taiso+gold.jpg)
![皇太子(明仁)による沖縄訪問 [1975年]](https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEjvSQrzV7yw_4gVQSwxZP_jh4VnEJscSqOqbiBh0VdAK3CRddXRqkd70JdLyws9fGejk-FGVmXWbHvSxlF3f8UogTyf9KXbqU1NGXesvcx2Hlsd6uq81AHweeioc61wynq3d2IYuyolijgT/s1600/akihito+message.jpg)

























































































































































































![アルニカ [ウサギ菊]](https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEilqv0qou-4NpoUh1PFWYK0FSaozKazee0VYGxsFtfjBma46ya9yxqB6X9Ziuob25tNRpBbnFIcUFlOEjz1WcAjVNzjGl1E-QbDgE7VOLkjZDx0eplJ1WJHf0fTEWXxf8F5G-cHUhqHELY9/s1600/ArnicaS.jpg)








































































































































.jpg)


















































































.jpg)